by Dr. Mark Proctor
The human brain is one of the most complex things known to mankind, and it requires a complex structure to house and protect it. Because our brains are proportionally quite large, and because we walk on two legs and therefore have a narrower pelvic outlet than four legged animals, our brain must emerge at an earlier stage in development than any other mammal. In order for us to be able to ultimately gain the intelligence and skills we have as adults, the brain must grow a great deal after birth, especially in that first year of life before we learn to walk and talk. It is the complex skull that allows for this growth to occur.
The cranium is composed of 5 major bones separated by 6 major seams called sutures. The early closure of any of these sutures, which occurs in one out of approximately every 2,200 births, can lead to abnormal growth patterns of the head as the brain expands in ways it was not meant to. This condition, called craniosynostosis, can lead to an abnormal shape of the head, and in some children it can even affect normal brain development. For this reason, surgical repair of this condition is often recommended.
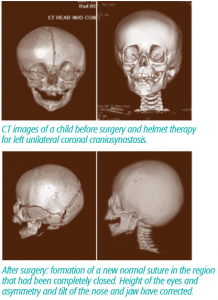 The methods for repairing craniosynostosis have come full circle over the years. The early operations involved a large incision, often extending from ear to ear, followed by release of the bones. However, this approach had a high failure rate, as many children fused the bones back together before the abnormal shape had corrected. This lead to the development of large cranial vault reconstruction operations, where a team of Plastic and Neurological Surgeons would remove and replace the cranial bones into a normal position. The advent of “minimally invasive techniques” using small incisions and endoscopes has brought the repair back to the early days of simply releasing the bones. Now the operations are highly successful because devices like helmets or internal springs direct growth in the proper orientation, and prevent the bones from fusing before things have corrected. This method takes advantage of the rapid brain growth that occurs very early in life. The optimal age for surgical intervention is before 3-4 months of age, as opposed to the larger operations which are frequently performed at 9-11 months of age.
The methods for repairing craniosynostosis have come full circle over the years. The early operations involved a large incision, often extending from ear to ear, followed by release of the bones. However, this approach had a high failure rate, as many children fused the bones back together before the abnormal shape had corrected. This lead to the development of large cranial vault reconstruction operations, where a team of Plastic and Neurological Surgeons would remove and replace the cranial bones into a normal position. The advent of “minimally invasive techniques” using small incisions and endoscopes has brought the repair back to the early days of simply releasing the bones. Now the operations are highly successful because devices like helmets or internal springs direct growth in the proper orientation, and prevent the bones from fusing before things have corrected. This method takes advantage of the rapid brain growth that occurs very early in life. The optimal age for surgical intervention is before 3-4 months of age, as opposed to the larger operations which are frequently performed at 9-11 months of age.
Endoscopic surgery can be used for any type of fused suture. It is more successful in single suture synostosis than in the more complex forms, in which the children are quite prone to re-fuse the cranial bones. The operations are relatively quick, taking about 30-40 minutes and a total of 2 hours in the operating room. The recovery is rapid with just an overnight hospital stay. The risk of a blood transfusion is under 5%. The child is fit with a helmet within a week after surgery, and requires helmet therapy for several months, possibly until their first birthday. The correction requires patience, as it takes at least 3-6 months to occur, but gradually the head improves to a normal shape. The risk of needing an additional operation is as low as 1-2% for non-syndromic single suture synostosis.
In summary, there are two surgical options for correction of craniosynostosis when it is diagnosed early in life. The traditional cranial vault reconstruction remains a mainstay for older children. Endoscopic suture release and post-operative helmeting is an excellent alternative for children if undertaken in the first 3-4 months of life.
Dr. Mark Proctor is Boston Children’s hospital’s Neurosurgeon-in-Chief; Director of Brain Injury Center, Professor at Harvard Medical School. He specializes in treatment of pediatric craniofacial abnormalities, spinal disorders, sports-related injuries and trauma to the brain and spine.